Investigadores del ISCIII acaban de publicar un estudio que ha analizado la evolución y diversidad genética del coronavirus SARS-CoV-2 en España al inicio de la pandemia.
Los resultados se publican en la revista ‘Journal of Virology’ y permiten establecer un ‘mapa’ de la transmisión geográfica de las diferentes variantes genéticas del virus, una información que ayuda a explicar la dispersión del SARS-CoV-2 y su evolución a lo largo de la pandemia.
Los autores de la investigación pertenecen a la Unidad de Virus Respiratorios y Gripe, la Unidad de Inmunopatología del Sida y la Unidad de Biología y Variabilidad del VIH, todas ellas pertenecientes al Centro Nacional de Microbiología (CNM), y las unidades de Genómica y Bioinformática del ISCIII. Según concluyen, la transmisión temprana del SARS-CoV2 en España se debió a un ‘efecto fundador’.

De esta manera, las variantes del SARS-CoV-2 que llegaron primero a España fueron las que se impusieron en los inicios de la circulación del virus, lo que explicaría la alta frecuencia de variantes del clado denominado en la literatura como 19B. Los clados son grupos o ‘familias’ de virus que comparten su historia evolutiva y que poseen un antepasado común.
A medida que el virus circulaba surgieron variantes virales que contenían una mutación en la posición 614 de la espícula del virus (D614G), que fueron sustituyendo a las variantes iniciales que carecían de esta mutación y que llegaron a imponerse gracias a la ventaja evolutiva que supone y que puede que les otorgue una mayor capacidad infectiva.
El estudio explica que las variantes genéticas más frecuentes al principio de la epidemia acabaron siendo sustituidas por otras variantes virales caracterizadas por presentar la mutación D614G, que se asocia a mayor capacidad de trasmisión. Esta mutación en la proteína de la espícula del virus le dota de una ventaja evolutiva, lo que le ha permitido imponer su circulación a lo largo del tiempo y llegar a ser la variante dominante.
El primer objetivo de la investigación ha sido analizar la diversidad genética y la distribución viral observada en los primeros 12.500 genomas de SARS-CoV-2 publicados en la base de datos GISAID, procedentes de todos los países que hasta el momento habían secuenciado el virus y entre los que se incluían un total de 290 genomas de España procedentes de 11 comunidades autónomas diferentes. 61 de estos genomas fueron secuenciados en el ISCIII y procedían de virus presentes en muestras respiratorias recibidas en el CNM entre la última semana de febrero y la primera de marzo, según explican los investigadores.
Francisco Díez, María Iglesias-Caballero y Javier García Pérez son los primeros firmantes del estudio e Inmaculada Casas y José Alcamí son los autores senior. Sara Monzón, Pilar Jiménez, Sarai Varona, Isabel Cuesta, Ángel Zaballos, Mercedes Jiménez, Laura Checa, Francisco Pozo, Mayte Pérez-Olmeda y Michael M Thomson han trabajado en diferentes aspectos de la investigación.
Cambios en las variantes predominantes
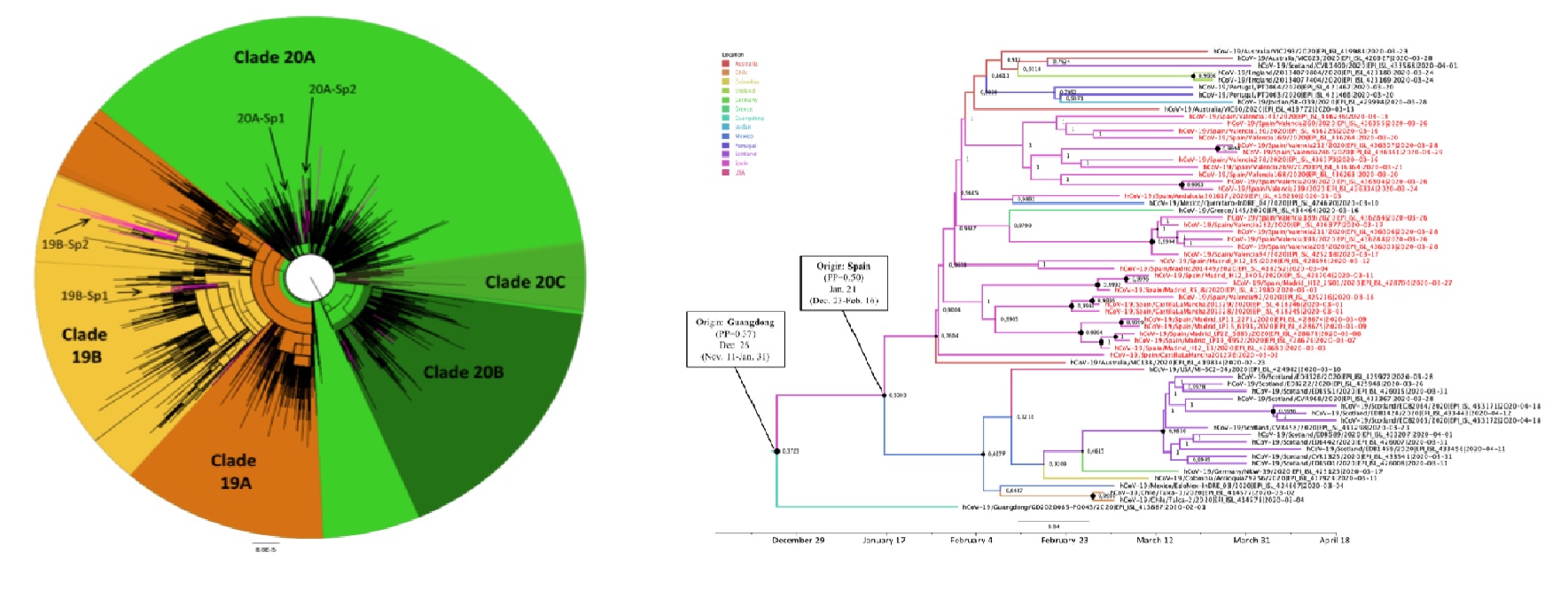
En su trabajo se ha detectado al menos 62 introducciones de diferentes variantes de SARS-CoV-2 en España a lo largo de los primeros meses de la pandemia. La mayoría de estas variantes se agruparon en tres clados denominados en la literatura como 20A, 20B y 20C, caracterizados por la presencia de la sustitución D614G en la proteína de la espícula (en el 56% de los casos) y en el clado 19B, diferenciado por la sustitución L84S en el gen que codifica la ORF8 (40% de los casos). Estas ‘señas de identidad genética’ de las variantes pueden definir cambios antigénicos del virus que suponen ventajas e influyen en su transmisibilidad.
La alta prevalencia de variantes del clado 19B durante las primeras semanas de circulación es una de las características principales del inicio de la epidemia en España. Sin embargo, esta situación cambió drásticamente a partir de mediados de marzo, cuando la mutación D614G fue imponiéndose en los virus circulantes; al final de ese mes llegó a ser la variante mayoritaria o dominante, una situación que se mantiene a día de hoy. De hecho, desde finales del mes de junio sólo se secuencian variantes D614G. El segundo objetivo de este trabajo consistió en el seguimiento de esta mutación a lo largo de toda la epidemia en España y se ha realizado mediante un segundo análisis de los primeros 4.242 genomas secuenciados en España.
Los científicos del Centro Nacional de Microbiología también han comprobado si la mutación D614G confiere algún tipo de ventaja evolutiva al virus. Para ello, han realizado experimentos ‘in vitro’ con pseudovirus que expresan la proteína de la espícula con y sin la citada mutación, y han obtenido resultados que apoyan una posible mayor infectividad asociada a la variante D614G. Esta mayor capacidad infectiva en esta mutación es una de las hipótesis que podrían explicar la sustitución de las variantes más antiguas, que carecen de dicha mutación, observada en España a lo largo del inicio de la epidemia de SARS-CoV-2.
Cuatro clusters de transmisión local entre enero y febrero
Por otro lado, la investigación ha determinado que cuatro de las 62 variantes introducidas de SARS-CoV-2 detectadas en España dieron lugar a cuatro clusters de transmisión local, que incluían un total de 110 secuencias diferentes del virus. Los análisis filodinámicos realizados al respecto permitieron caracterizar el origen geográfico y temporal de estas 4 variantes y situarlas en España entre finales de enero y principios de febrero del 2020, lo que confirma que el virus producía infecciones puntuales varias semanas antes del gran aumento de casos detectado a principios de marzo.
En concreto, dos de estas variantes presentaban la mutación L84S en el gen de la ORF8, y las otras dos, la mutación D614G en el gen de la espícula. Todo ello sugiere que el inicio de la epidemia en España estuvo marcado por un ‘efecto fundador’, con protagonismo de las variantes que carecían de la mutación D614G. En una etapa posterior, estas variantes que se habían expandido ampliamente al principio de la epidemia fueron completamente sustituidas por variantes D614G, debido a la citada ventaja evolutiva que parece otorgar dicha mutación.